We also need to understand the concept of protease inhibitor boosting. Kaletra is a “boosted”
protease inhibitor, meaning it combines the active drug lopinavir with a low dose of ritonavir,
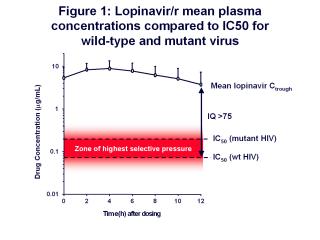
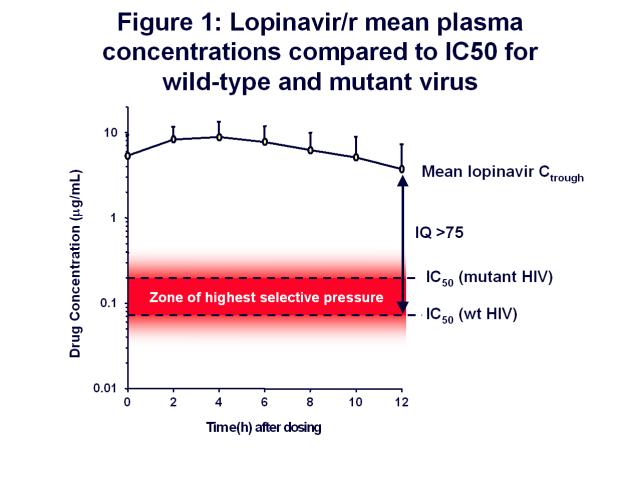
which is used to boost the levels of lopinavir in the patient’s blood. If you look at a plot of
lopinavir plasma levels over time (see figure 1), the concentration of lopinavir is well
above the IC50 for wild-type virus during the 12-hour dosing interval when given as
Kaletra. The IC50 value for mutant virus is higher, and defines the upper limit of a
“zone of highest selective pressure.” If drug concentrations fall to levels within this zone,
viral replication will lead to the rise of viral mutants through selective drug pressure. Drug
concentrations below this zone will allow preferential replication of the wild-type virus, and
concentrations above will suppress all replication. It is the drug concentrations within this
zone that will suppress wild-type replication but will allow viral mutants to escape.
The longer drug concentrations remain in this zone, the more you will favor the replication of the
mutant virus because you are inhibiting the majority of wild-type virus but the minority of the
mutant virus. The width of this zone varies by drug, and for drugs such as 3TC and efavirenz, the
zone is large. Resistance comes very easily to those drugs because the drug concentrations
remain in this zone for relatively longer periods of time.
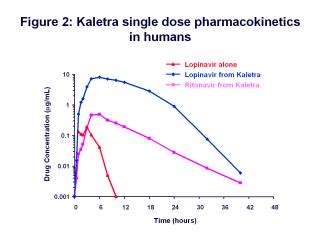
We believe that with Kaletra, drug levels don't reside in the zone of selective pressure for very
long, and consequently there is little resistance development. With other drugs, such as nelfinavir,
that reside longer in this zone, more resistance develops. Let's look at our graph and assume that
one or two or more doses of Kaletra are missed, and then ask what happens to the
pharmacokinetics of lopinavir?
|

{kind=link}
{kind=link}